Experts discuss recent advances in cell viability testing methods in bioreactors.
Quality/GMPs
Latest News
Advertisement
Digitalization of QbD Risk Assessments
The digital transformation of quality-by-design assessment workflows can improve efficiency, reduce human errors, and facilitate integration within a much broader digital ecosystem.

Subjectivity in Quality Risk Management
The authors discuss subjectivity in the ICH Q9 (R1) guidance document.

Phase-appropriate Compliance for Cell and Gene Therapies
Understanding how to apply phase-appropriate GMPs is crucial for achieving successful regulatory approval.
Advertisement

Quality, flexibility, and cost savings are driving use of perfusion technology in biosimilars manufacturing.
Multiple methods are required for detecting and removing protein impurities.
Regulators and manufacturers address economic and ethical issues for scarce medicines.
Susan Schniepp, Distinguished Fellow at Regulatory Compliance Associates, discusses the regulatory requirements for improving manufacturing lines.

Industry experts discuss the challenges of using single-use systems in biopharma manufacturing.
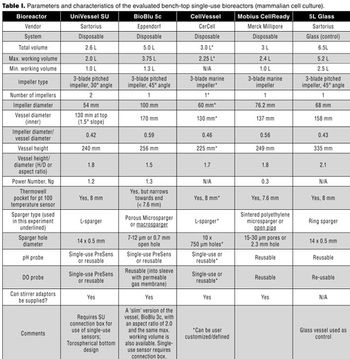
The authors present a case study in which four single-use vessels were fitted to an existing bioreactor system.
The study demonstrates a systematic approach to stabilize PBS-formulated mAbs against freeze-thaw degradation.
FDA and industry seek speedy Congressional approval of new user fee plan.
The mAb, in combination with lenalidomide and dexamethasone, or bortezomib and dexamethasone, was granted a Breakthrough Therapy Designation from FDA for the treatment of multiple myeloma.
The companies filed a BLA for romosozumab, an investigational monoclonal antibody for the treatment of osteoporosis.
The agency will review Mylan and Biocon’s biosimilar to pegfilgrastim.
The company manufactures biological drug products and intermediates for the allergy vaccine market.
The agency recommends suspending drugs developed with bioequivalence studies performed at Semler Research Centre Private Ltd, Bangalore, India.
GenPak Solutions is cited by FDA for cGMP violations at its Hilliard, Ohio facility.
The committee voted unanimously in favor of approving the drug, but the majority supported implementing additional risk management.
FDA issued a warning letter to the Worthing, UK facility for cross contamination and microbial contamination cGMP violations.
The agency says the increasing requests for orphan drug designation has resulted in a change in FDA’s review goals.
The agency says that the routine large-scale compounding of drugs that are exact copies of existing medications undermines the the drug approval process.
The draft guidance addresses control of elemental impurities in harmonization with implementation of ICH Q3D guideline.
FDA cited Guangzhou Haishi Biological Technology Co., Ltd. with CGMP violations.
The agency says, for now, it’s business as usual.The European Medicines Agency (EMA) says the future location of the agency will be determined by common agreement between representatives of the Member States, according to a July 6, 2016 statement. Until then, EMA says it will be conducting business as usual, and the outcome of the June 23 referendum will not affect the agency’s operations.
Revised versions of ISO 14644 Parts 1 and 2 introduce changes to sampling procedures and monitoring plans for cleanrooms and clean zones.
The author provides a review of the concepts of design and qualification that apply to single-use systems.
A streamlined approach may enhance process efficiency and product quality.
Advertisement
Advertisement
Trending on BioPharm International
1
How CDMO Alliances Can Provide End-to-End Service that Reduces Drug Development Time and Costs
2
FDA Clears PharmaResearch IND for Nano-Based Cancer Drug PRD-101
3
First-in-Human Study Validates Safety of Next-Generation mRNA–LNP Platform
4
High-Dose Nusinersen Slows Neurodegeneration in SMA Patients, Study Shows
5