The agency publishes draft guidance on best practices for communication between FDA and IND sponsors during drug development.
Quality/GMPs
Latest News
Advertisement
Digitalization of QbD Risk Assessments
The digital transformation of quality-by-design assessment workflows can improve efficiency, reduce human errors, and facilitate integration within a much broader digital ecosystem.

Subjectivity in Quality Risk Management
The authors discuss subjectivity in the ICH Q9 (R1) guidance document.

Phase-appropriate Compliance for Cell and Gene Therapies
Understanding how to apply phase-appropriate GMPs is crucial for achieving successful regulatory approval.
Advertisement
FDA confirmed quality focus while Congress moved to bolster biomedical innovation.

Expectations are high for rapid testing methods, but demonstration of comparability proves challenging.
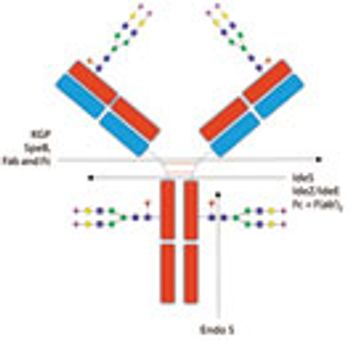
Advances in glycan analysis are enhancing biologics development and quality control processes.
Finalizing GMP requirements and quality standards for the development, manufacture, and clinical testing of ATMPs in the EU is proving to be a complex task.
The agency promotes safer use of drugs and prevention of medication errors through a new webpage and practice guide.
FDA seeks industry support for metrics program, emphasizing the surveillance focus.
There’s renewed optimism in the biomedical research community that years of effort finally may begin to pay off for developing cutting-edge gene and cellular treatments for debilitating and life-threatening conditions. Jill Wechsler reports.
Pharmaceutical manufacturers should not be protected from antitrust litigation simply because FTC chooses not to pursue a lawsuit, the agency wrote in a recent amicus brief.
Robert Califf addresses questions about drug pricing at the Senate hearing to weigh his appointment to be the next commissioner of FDA.
The new executive director of the European Medicines Agency begins appointment.
Panelists at the meeting will focus on clinical trial design, immunogenicity, and enhancing implementation plans for administering already-licensed vaccines to this patient population.
FDA seeks feedback on possible analytical standards and approaches to optimize regulation of next-generation sequencing (NGS)-based in vitro diagnostic tests.
Even though rising production and use of generic pharmaceuticals is saving billions for the nation’s healthcare system, policy makers continue to slap the industry with policies it claims will limit product development and sales.
The European agency presents guidelines for conducting post-authorization efficacy studies.
All biosimilars for a specific product will be reimbursed with the same J-code under Medicare Part B regardless of manufacturer, according to a CMS rule that was proposed in July 2015 and finalized on Oct. 30, 2015. The rule was finalized prior to any formal guidance from FDA on interchangeable products. CMS said it did not consider interchangeability into its decision, as there are no currently approved interchangeable biologics on the market.
Diversifying the Global Heparin Supply Chain: Reintroduction of Bovine Heparin in the United States?
The global supply chain for bovine and porcine heparin and regulatory considerations are examined.
New program emphasizes quality, risk, and global collaboration.
Siegfried Schmitt, principal consultant, PAREXEL, discusses how to ensure archive records can be retrieved.
The authors discuss performing investigations of biological products.

Virtual pilot programs examine scenarios that may occur while implementing serialization requirements for the US Drug Supply Chain Security Act.
In 2016, Amgen will simultaneously fight off biosimilar competitors to its legacy products and prepare to file its own follow-on products with regulators.
The agency issued a draft guidance document on the requirements for submission of applications for liposome drug products.
USP responds to FDA's draft guidance on the naming of biological products.
Despite considerable investment by biotech manufacturers in developing competitive biologics for the US market, gaining FDA approval of these products has turned out to be a slow and complex process.
Advertisement
Advertisement
Trending on BioPharm International
1
How CDMO Alliances Can Provide End-to-End Service that Reduces Drug Development Time and Costs
2
FDA Clears PharmaResearch IND for Nano-Based Cancer Drug PRD-101
3
First-in-Human Study Validates Safety of Next-Generation mRNA–LNP Platform
4
High-Dose Nusinersen Slows Neurodegeneration in SMA Patients, Study Shows
5